

-
Density functional theory study of homologous organometallic molecules of the [RhXL2]2 (X=Cl, Br, or I); L=CO, PH3, or PF3) type.
P. Seuret, J. Weber and T.A. Wesolowski
Molecular Physics, 101 (16) (2003), p2537-2543


DOI:10.1080/0026897031000112497 | unige:3502 | Abstract | Article PDF
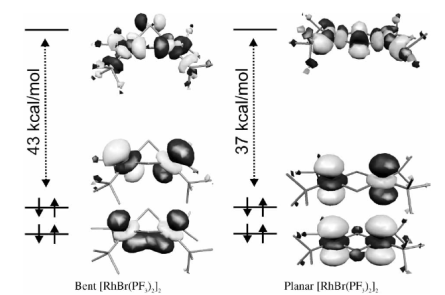
Density functional theory generalized gradient approximation calculations, which were tested in our previous detailed study of [RhCl(PF3)2]2 (Seuret et al., 2003, Phys. Chem. chem. Phys., 5, 268-274), were applied for a series of homologous organometallic compounds of the [RhXL2]2 (X = Cl, Br, or I; L = CO, PH3, or PF3) type. Various properties of the studied compounds were obtained. Optimized geometries of [RhCl(PH3)2]2 and [RhCl(CO)2]2 are in very good agreement with available experimental data. Geometries of other compounds as well as other properties (thermochemistry of selected fragmentation channels, barriers to structural changes, frontier orbitals) which are not available experimentally were predicted. All the considered compounds are not planar. Enforcing planarity of the central [RhX]2 moiety requires only a small energetic cost ranging from 2.2 to 3.9 kcal mol-1. The analysis of frontier orbitals indicates that the metals provide the most favourable site for the electrophilic attack in all considered compounds. The analysis of the shape of the lowest unoccupied molecular orbitals indicates that the halogens and ligands provide the most favourable site for the nucleophilic attack for [RhCl(CO)2]2 or [RhCl(PF3)2]. For [RhBr(PF3)2]2, [RhI(PF3)2]2 and [RhCl(PH3)2]2, the nucleophilic attack on the halogen is less probable. Except for [RhCl(CO)2]2, the least energetically expensive decomposition channel involves initial separation of ligands. For [RhCl(CO)2]2, its decomposition into the RhCl(CO)2 fragments was found to be the least energetically expensive fragmentation reaction which is probably one of the reasons for the known catalytic activity of this compound.